Контрактные исследовательские организации
Контрактная исследовательская организация (Contract Research Organization, CRO) – это коммерческая научно-исследовательская компания, которая в рамках договора со спонсором предоставляет специализированные услуги по управлению клиническими исследованиями для фармацевтической, биотехнологической и медицинской промышленности .
Основная роль контрактной исследовательской организации заключается в планировании, координации, выполнении и контроле процессов, связанных с проведением клинического исследования. Контрактная исследовательская организация может являться посредником между спонсором исследования и другими участниками исследования (например, комитетами по этике, регулирующими органами, поставщиками и медицинскими учреждениями).
Какие задачи решает контрактная исследовательская организация?
- доклинические исследования;
- клинические исследования;
- пострегистрационные исследования;
- обработка данных и статистика;
- оформление документации;
- фармаконадзор.
Контрактные исследовательские организации являются ключевыми участниками клинических исследований, поскольку их специалисты обладают знаниями и возможностями, необходимыми для надлежащего проведения клинического исследования. Они обеспечивают качество проведения испытаний и соответствие национальным и международным стандартам.
Специалисты контрактных исследовательских организаций – это высококвалифицированные научные сотрудники, имеющие ученую степень в области медицины, биологии или фармацевтики .
Специалист по клиническим исследованиям (Clinical Research Associate, CRA), также называемый монитор клинических исследований или сокращенно монитор, – это научный сотрудник, координирующий клинические испытания лекарств, биопрепаратов или медицинского оборудования. Специалисты по клиническим исследованиям организуют и проводят клинические испытания новых или уже зарегистрированных лекарственных средств с целью оценки их эффективности, безопасности и качества .
Основные обязанности монитора включают в себя :
- написание методических инструкций для проведения клинических испытаний лекарственных препаратов;
- поиск и инструктаж подходящих специалистов (врачей, исследователей) для проведения клинических испытаний;
- контроль проведения клинического исследования на всем их протяжении, включая регулярное посещение исследовательских центров, проверку данных пациентов в историях болезни и решение всех возникающих проблем;
- обеспечение исследовательских необходимым оборудованием и материалами для проведения исследования;
- написание технических отчетов.
Опыты на животных
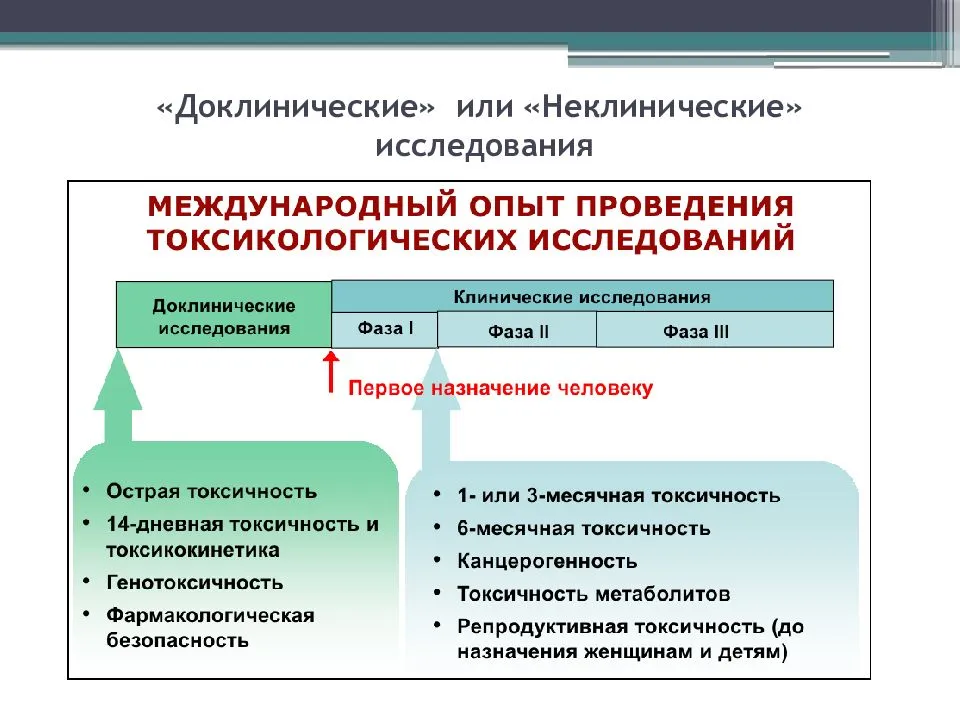
Доклинические испытания — это масштабный этап исследования кандидатов в лекарства. Он может длиться до 10 лет и проводится преимущественно на животных. Некоторые тесты, например при исследовании мутагенности, традиционно выполняют на культурах клеток, бактериях, мухах дрозофилах или аквариумных рыбках. В продвинутых лабораториях отдельные эксперименты научились моделировать в виртуальной реальности. Однако основные испытуемые — это грызуны: мыши, крысы, хомяки, морские свинки и кролики. Ряд тестов требует участия более крупных млекопитающих: собак, обезьян, реже — мини-пиги, кошек, хорьков и др.
Исследование проводится на здоровых животных обоих полов, отобранных по возрасту, размерам и происхождению. Испытуемых содержат в стандартизованных условиях, кормят и поят строго по режиму одинаковой пищей. Если будущее лекарство предназначено для детей, то исследования проводят на детенышах, лекарства для беременных испытывают на беременных самках, препараты геронтологической практики — на стареющих животных.
Цель доклинических исследований:
- Отсеять вещества, непригодные для дальнейших испытаний из-за токсичности, канцерогенности или других свойств.
- Определить «стартовую» безопасную дозу препарата для использования в первой фазе клинических исследований на людях.
- Оценить соотношение польза/риск для каждого кандидата в лекарства.
Важнейший аспект доклинических испытаний — проверка вещества на вредность. Начинают с определения дозы: нормальной переносимой, токсической и летальной. Определяют, какие органы и ткани животных оказываются более подвержены действию препарата? Какие побочные эффекты вызывает вещество?
Сначала препарат вводят с короткими 3-6-часовыми промежутками в течение суток и отслеживают результаты острой токсичности. Затем приступают к тестированию хронической токсичности. Эта часть исследования зависит от предполагаемой продолжительности лечебного курса у человека и может длиться от нескольких недель до года и более.
Далее испытывают влияние препарата на фертильность у самцов и самок животных. Определяют эмбриотоксичность и фетотоксичность — отрицательное влияние вещества на эмбрион, плод, а также отслеживают рост и развитие детенышей после рождения. Отдельно изучают мутагенность — способность вызывать мутации у взрослого животного и канцерогенность — вероятность злокачественных опухолей.
- Сведения о канцерогенности вещества обязательно необходимы для получения разрешения на клинические исследования в нашей стране. Лекарства, которые будут продаваться без рецепта или предназначенные для длительного использования тестируются с особой тщательностью. Например, гормональные препараты проходят тестирование на канцерогенность около 7-10 лет, и подопытными, помимо крыс и мышей, должны быть собаки или обезьяны.
Данные проверки на вредность являются опорными для последующих экспериментов. Существуют коэффициенты пересчета доз исходя из массы тела. Ориентируясь на эти цифры можно проецировать влияние препарата на человека.
Второй аспект доклинических исследований — фармакокинетика — путь, который проделывает вещество в организме: как всасывается, как распределяется, вступает ли в химические превращения, какими органами и с какой скоростью выводится. Знание фармакокинетики дает представление о достижимой концентрации вещества в крови, а значит, позволяет точнее подобрать эффективную дозу для человека, выбрать оптимальную форму введения препарата в организм: через рот или с помощью инъекций.
Третий вопрос, на который отвечают лабораторные изыскания — предположительная эффективность кандидата в лекарства. Методология этой части исследования значительно отличается в зависимости от целевой сферы применения вещества. Например, при тестировании нейролептиков у животных исследуют их поведенческие реакции и двигательную активность. Для тестирования сердечных лекарств проводят испытания на собаках, кошках или обезьянах, у которых искусственно нарушают кровообращение сердца, имитируя стенокардию.
Как узнать, как будет проходить конкретное клиническое исследование?
Метод, длительность, требования к участникам и другие подробности клинического испытания прописываются в протоколе исследования. Для безопасности пациентов протокол, разрабатываемый спонсором исследования, проходит экспертизу в Министерстве здравоохранения Российской Федерации, а именно в комитете по этике.
С протоколом исследования пациента должны ознакомить до начала тестирования. Если участника устраивают все условия, он подписывает информированное согласие, в котором подтверждает, что ознакомился со всеми документами и добровольно решил принять участие в тесте.
Цели клинических исследований [1]
- Во-первых, установить безопасность и переносимость лекарственных препаратов для здоровых добровольцев.
- Во-вторых, подобрать оптимальную дозировку препарата, длительность курса лечения для пациентов с определенным заболеванием. Если исследуется вакцина, то подбирается оптимальная доза и схема вакцинации иммунобиологическими лекарственными препаратами здоровых добровольцев.
- В-третьих, установить безопасность и эффективность препарата для пациентов с определенным заболеванием или профилактическую эффективность иммунобиологических лекарственных препаратов (вакцин) для здоровых добровольцев.
- Также во время клинических исследований изучается возможность расширения показаний препарата для медицинского применения и выявляются ранее неизвестные нежелательные реакции уже зарегистрированных лекарственных препаратов.
Развитие клинических исследований проходило по долгому и увлекательному пути – от изучения влияния питания на здоровье человека (VI век до н. э.) до первого рандомизированного контролируемого испытания стрептомицина в 1946 году. История клинических исследований к настоящему моменту охватывает широкий спектр проблем – научных, этических и нормативных .
- В 1747 году британский врач Джеймс Линд показал, что цитрусовые и зелень предотвращают заболевание цингой у моряков во время длительных экспедиций. Линд разделил команду корабля на несколько равных групп и оценил, как определенные продукты влияют на развитие цинги. Это открытие считают одним из первых зарегистрированных клинических исследований, которое проводилось с использованием научных методов .
- Спустя век наступает еще одна важная веха в истории современных клинических испытаний – возникновение понятия плацебо. В 1811 году в Медицинском словаре Хупера появляется следующее определение плацебо – это «эпитет, данный любому лекарству, которое приносит больше удовлетворения для пациента, чем пользы». В 1863 году американский врач Остин Флинт проводит первое клиническое исследование, сравнивающее фиктивное лекарство (плацебо) с активным лечением при ревматизме .
- Первое двойное слепое контролируемое клиническое исследование было проведено в 1943 году Советом по медицинским исследованиям Великобритании для изучения действия патулина при простуде. В ходе эксперимента ни испытуемые, ни врачи не знали, получают ли они настоящий препарат .
- Первым рандомизированным контролируемым испытанием считается изучение стрептомицина при туберкулезе легких, проведенное в 1946 году Советом по медицинским исследованиям Великобритании. Пациенты с туберкулезом случайным образом были распределены в две группы. Ни участники испытания, ни исследователи, осуществлявшие анализ результатов, не знали о том, к какой группе отнесен тот или иной больной. В экспериментальной группе испытуемые соблюдали постельный режим и получали стрептомицин, а в контрольной группе – только соблюдали постельный режим. Эффективность лечения туберкулеза оценивалась в сравнении с контрольной группой. Это исследование стало главной моделью изучения лекарственных препаратов, а рандомизированное распределение используется в клинических испытаниях и в настоящее время .
- Важным этапом развития клинических исследований стала Талидомидовая трагедия». Лекарственный препарат талидомид был очень популярным снотворным и успокоительным средством, особенно у беременных женщин. К сожалению, проведенных лабораторных экспериментов до регистрации и начала продаж препарата было недостаточно – исследователи не обнаружили тератогенные свойства талидомида, он вызывал тяжелые аномалии развития плода. Препарат был запрещен для широкого использования в большинстве стран в 1961 году. Этот печальный опыт послужил основой для проведения более тщательных доклинических исследований лекарств .
- В середине XX века возникла необходимость регулировать и контролировать проводимые клинические испытания с точки зрения этики и защиты прав пациента. Важными нормативными документами являются Нюрнбергский кодекс (1947 г., впервые утвердил строго добровольное участие в клинических исследованиях), Хельсинкская декларация, Бельмонтский доклад и Руководство по надлежащей клинической практике (GCP) .
Регуляторы всех стран, объединяйтесь!
Первые руководства и законы, разработанные в разных государствах для регуляции КИ, неизбежно различались между собой. В 1980-х годах страны Европы начали создавать единое экономическое пространство, которое позднее легло в основу Евросоюза. В рамках данного процесса необходимо было сформировать общий рынок лекарственных средств. Принятие единых требований к регистрации и обращению лекарств позволило бы избежать дублирования исследований и избыточных бюрократических процедур. Конкретные планы по унификации были названы в 1989 году на Международной конференции регуляторных органов в сфере обращения лекарственных средств, организованной ВОЗ (WHO International Conference of Drug Regulatory Authorities). В результате в 1990 году представители не только стран Европы, но и США и Японии создали Международный совет по гармонизации технических требований к фармацевтическим препаратам для человека (International Council for Harmonisation, ICH).










ICH стал организацией, объединившей регуляторные органы разных стран и фармацевтическую промышленность, чтобы совместными усилиями создать универсальные международные научные и технические требования к разработке лекарств, проведению КИ, производству и обращению препаратов. В настоящее время участниками ICH являются страны Евросоюза, США, Япония, Канада, Бразилия, Китай и другие государства. Россия и страны ЕАЭС выступают в качестве наблюдателей.
ICH разработал множество руководств, которые поделены на четыре категории: качество, безопасность, эффективность и мультидисциплинарные руководства. Ознакомиться с ними можно на официальном сайте организации. Основная часть руководств ICH, посвященных эффективности и безопасности лекарственных средств, имеет непосредственное отношение к проведению КИ. Они рассматривают этические и научные вопросы, статистические методы и дизайн исследований
Особое внимание уделяется изучению токсичности лекарств
Основной международный стандарт проведения КИ – «Надлежащая клиническая практика» (Good Clinical Practice, GCP) является одним из руководств, созданных ICH.
Первая версия GCP R1 была опубликована в 1996 году. В ней были описаны права и обязанности всех участников КИ, включая исследователей, мониторов (контролеров), представителей спонсора и этических комитетов, а также требования к документированию исследования. В 2016 году вышло руководство GCP R2 – дополненная версия, освещающая методы более эффективного проведения исследований и вопросы электронного документооборота.
Протокол планируемого КИ – цели, дизайн, число участников – формируется исходя из стратегии фармацевтической компании с учетом регуляторных требований стран, где планируются продажи разрабатываемого лекарства. С целью одновременной регистрации в нескольких государствах организуются международные многоцентровые КИ. В таких исследованиях изучение препарата происходит одновременно в нескольких странах по единому протоколу.
Как упоминалось в нашей первой статье, посвященной КИ, при создании документов, регулирующих сферу КИ в России и ЕАЭС, за основу были взяты международные рекомендации, разработанные ICH. Однако пока что в нашей стране официально лишь часть руководств соответствует требованиям ICH. Это различие осложняет вывод российских препаратов на международные рынки, так как отечественные исследования зачастую не соответствуют международным требованиям.
Чтобы наглядно продемонстрировать разницу в требованиях к КИ для регистрации лекарств в России и в Европе, можно сравнить, к примеру, как исследовались оригинальные препараты с одинаковым механизмом действия. Гозоглиптин и вилдаглиптин – ингибиторы ДПП-4, предназначенные для контроля уровня глюкозы при сахарном диабете II типа. Вилдаглиптин был разработан швейцарской компанией Novartis и одобрен Европейским медицинским агентством (European Medicines Agency) в 2007 году, после завершения КИ с участием в общей сложности около 4500 пациентов. Гозоглиптин был изначально разработан американской компанией Pfizer, которая провела КИ I и II фазы с участием приблизительно 600 пациентов и свернула испытания. Затем лицензия на препарат была передана российской компании «Сатерекс», которая провела успешное КИ III фазы с участием 299 пациентов, где сравнивалась эффективность гозоглиптина и вилдаглиптина, и на основании собранных данных получила регистрационное удостоверение в России в 2016 году. В Европе и США данный препарат не зарегистрирован.
Опасные заблуждения
За последние годы сформировался ряд опасных заблуждений о законности офф-лейбл. Пройдемся же по ним тяжелой юридической пятой.
| Заблуждение | Правовая реальность |
| Режимы офф-лейбл вошли (со значком #) в клинические рекомендации, следовательно они законны. Отсутствие препаратов, применяемых офф-лейбл, в стандартах медицинской помощи не критично, так как последние используются для экономических целей, а для врача главным документом служат клинические рекомендации | Несмотря на то, что режимы офф-лейбл вошли в клинические рекомендации нового поколения в соответствии с приказом Минздрава от 28.02.2019 №103н*, это не сделало их использование законным, так как дальнейшей системной регламентации норм не последовало. Именно поэтому схемы офф-лейбл и не вошли в стандарты медицинской помощи (которые, согласно закону**, разрабатываются на основе рекомендаций), ведь в отличие от рекомендаций стандарты являются нормативно-правовыми актами, обязательными к применению. И никакая особая «узкоэкономическая» роль стандартов законодателем не определена. Базовый федеральный закон №323-ФЗ***, Порядок назначения лекарственных препаратов**** и иные акты в совокупности не допускают использование препаратов офф-лейбл. У пациентов младше 18 лет применение препаратов офф-лейбл станет допустимым с 1 июля 2022 года – после вступления в силу федерального закона №482-ФЗ и его подзаконных актов – специально принятых с целью легализации офф-лейбл. Только после этого станет возможно и их включение в стандарты медпомощи |
| Режимы офф-лейбл попали в КСГ. Оплата назначений офф-лейбл в системе ОМС говорит о законности такой деятельности | Оплата назначений офф-лейбл в системе ОМС не говорит о законности такой практики, так как КСГ является системой тарификации медицинской помощи и не регулирует порядок использования лекарственных препаратов |
| Назначения офф-лейбл законны на основании решения ВК | ВК не имеет подобных полномочий. Практика подобных назначений свидетельствует разве что о превышении ВК своих полномочий. Порядок создания и деятельности ВК*****, Порядок назначения лекарственных препаратов****** и иные нормативные правовые акты не содержат указаний на подобные функции ВК.          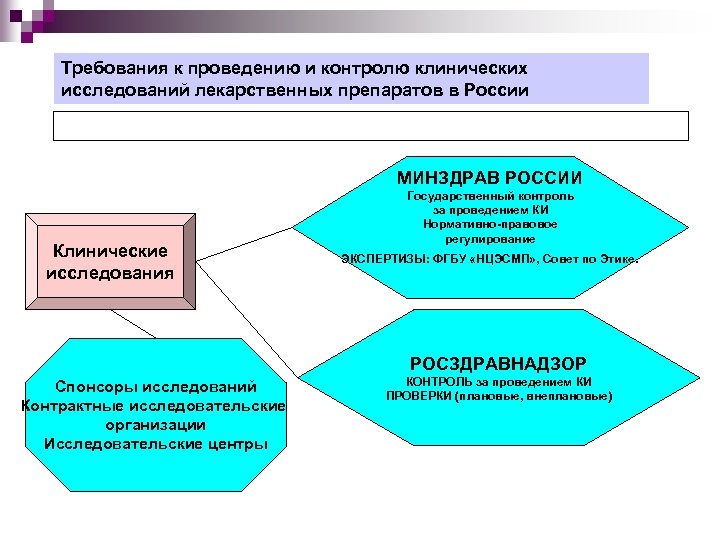 Нередко за полномочие ВК назначать офф-лейбл ошибочно принимается право ВК назначать препараты, не входящие в стандарт медпомощи или не предусмотренные клиническими рекомендациями, в случае индивидуальной непереносимости или по жизненным показаниям (п. 15 ст. 37 федерального закона №323-ФЗ) |
| Запрещено использование незарегистрированных лекарственных препаратов (офф-лейбл), а использование зарегистрированных препаратов даже не в соответствии с инструкцией допустимо | Офф-лейбл – это применение зарегистрированных лекарственных препаратов не в соответствии с инструкцией. Видов таких назначений более десяти, в том числе использование препарата по показаниям, не указанным в инструкции; без учета противопоказаний, указанных в инструкции; использование препарата в дозах, по схеме, в комбинации, в режиме или по иным параметрам, отличающимся от указанных в инструкции и др. Применение незарегистрированных препаратов не входит в понятие офф-лейбл |
| Инструкции к препаратам не обязательны, так как не являются нормативными актами | Инструкция по медицинскому применению лекарственного препарата обязательна к соблюдению. Она обладает свойствами нормативного характера, так как входит в состав регистрационного досье, согласовывается с Минздравом России в ходе государственной регистрации препарата и выдается одновременно с регистрационным удостоверением. Изменение инструкции, в том числе сведений о показаниях к применению препарата, требует проведения новых клинических исследований и экспертизы качества препарата. Это позволяет квалифицировать использование офф-лейбл как нарушение критериев качества и безопасности медицинской помощи |
| Раз нет четкого запрета на применение офф-лейбл, следовательно оно допустимо | Совокупность норм права свидетельствует о недопустимости назначений офф-лейбл, за исключением такого использования препаратов у пациентов до 18 лет, которое станет допустимым с 1 июля 2022 года |
Какие бывают виды клинических исследований?
Особенности проведения клинических испытаний также зависят от типа исследований.
- Открытые исследования. При открытом исследовании, которое направлено только на изучение свойств препарата, без сравнения с другими уже известными лекарствами, и врач и пациенты осведомлены о том, какое лекарство проверяют;
- Сравнительные исследования. Этот тип исследования необходим для того, чтобы сравнить новый препарат с уже существующими методами лечения. Для этого всех участников делят на две группы: контрольная (получающая уже проверенное стандартное лекарственное средство) и тестовая (проходящая терапию новым препаратом;
- Слепые исследования. Слепые исследования — противоположность открытым. При простом слепом испытании только пациент не осведомлен, какое лечение он получает. При двойном слепом — это остается в тайне и от врача и от пациента. Таким образом стараются снизить риск подтасовки результатов теста из-за заинтересованности кого-то из участников исследования в получении определенных результатов;
- Плацебо-контролируемые исследования. Плацебо — препарат «пустышка», который получает часть пациентов, тогда как вторая часть принимает настоящее лекарство. Плацебо по виду и вкусу не отличаются от тестируемого препарата, чтобы участники не смогли догадаться, что проходят «ненастоящую» терапию. Такой метод позволяет оценить, какую роль в результатах теста занимает психологическая составляющая: внушаемость пациентов, внимательное отношение врача и сам факт лечения.
Как стать участником клинического исследования?
Для участия в клинических исследованиях добровольцы должны соответствовать определенным критериям. Критерии могут быть основаны на различных факторах, основные из которых – возраст, пол, история болезни, текущее состояние здоровья, прием лекарств и аллергии. Для некоторых исследований необходимы пациенты с определенным заболеванием или состоянием, в то время как другие исследования нуждаются в здоровых добровольцах.
Факторы, которые позволяют вам участвовать в клиническом исследовании, называются критериями включения, а факторы, которые не допускают участие, называются критериями исключения
Важно отметить, что критерии включения и исключения используются для определения подходящих участников и обеспечения их безопасности. Эти критерии помогают гарантировать, что исследователи смогут ответить на вопросы, которые они планируют изучить
Если пациент хочет принять участие в клиническом исследовании, он может подать заявку с помощью лечащего врача или самостоятельно. Для поиска клинических исследований, проводимых в России, можно воспользоваться следующими интернет-ресурсами:
- Официальный реестр разрешений на проведение клинических исследований (РКИ) Минздрава России. URL: https://grls.rosminzdrav.ru/CIPermitionReg.aspx.
- Международный реестр клинических исследований Национального института здоровья США http://www.clinicaltrials.gov.
- Интернет-сайты фармацевтических компаний.
В состав группы клинических исследований входят врачи и медсестры, а также социальные работники и другие медицинские работники. Они проверяют состояние здоровья участника в начале исследования, дают конкретные инструкции по участию в исследовании, внимательно следят за пациентом во время исследования и поддерживают связь после завершения исследования .
Согласно Федеральному закону от 12.04.2010 N 61-ФЗ (ред. от 13.07.2020) «Об обращении лекарственных средств» участие в клиническом исследовании является добровольным. Испытуемый может отказаться от участия в исследовании в любое время. Медицинское состояние пациента тщательно контролируется на протяжении всего исследования опытным научным персоналом и врачами. Для каждого исследования установлены параметры безопасности .
Информированное согласие – это обязательное требование медицинской этики и права, которое гласит, что пациенту должна быть предоставлена полная информация об исследовании, прежде чем он примет решение принять участие. Каждому кандидату на участие в исследовании выдается форма информированного согласия, в которой подробно описывается, что будет, если вы участвуете в исследовании. В письменной форме пациент получает следующую информацию :
- о лекарственном препарате для медицинского применения и сущности клинического исследования этого лекарственного препарата;
- о безопасности лекарственного препарата для медицинского применения, его ожидаемой эффективности и степени риска для пациента;
- об условиях участия пациента в клиническом исследовании лекарственного препарата для медицинского применения;
- о цели или целях и продолжительности клинического исследования лекарственного препарата для медицинского применения;
- о действиях пациента в случае непредвиденных эффектов влияния лекарственного препарата для медицинского применения на состояние его здоровья;
- об условиях обязательного страхования жизни и здоровья пациента;
- о гарантиях конфиденциальности участия пациента в клиническом исследовании лекарственного препарата для медицинского применения.
Если пациент решит принять участие в исследовании, то ему будет предложено подписать информационный листок пациента, что является подтверждением добровольного согласия пациента на участие в клиническом исследовании .
Рынок клинических исследований
Количество проводимых клинических исследований постоянно растет. Так в 2019 году Минздрав России выдал 746 разрешений на клинические исследования, что на 14,2% превышает показатель 2018 года. Всего в России на 2020 год проходит 5125 клинических исследований .








Наибольшее число клинических исследований проводится в Соединенных Штатах Америки – около 135000 зарегистрированных испытаний на 2020 год. Лидерами по числу проводимых клинических испытаний среди стран также являются :
- Франция (26178);
- Канада (22371);
- Германия (20275);
- Великобритания (18993);
- Китай (18628);
- Испания (14241);
- Италия (14109);
- Южная Корея (11332);
- Бельгия (10144).
В России в 2019 году наибольшее число международных многоцентровых клинических исследований было проведено в следующих терапевтических областях :
- онкология и онкогематология (29,1%);
- неврология (10,5%);
- ревматология (9,3%);
- психиатрия (6,7%);
- гематология и инфекционные заболевания (5,8%).
Среди локальных исследований и исследований биоэквивалентности дженериков и биоаналогов иностранных спонсоров наибольшее число клинических исследований по :
- эндокринологии (19,2%);
- кардиологии и сердечно-сосудистым заболеваниям (18,3%);
- офтальмологии (6,7%);
- урологии (6,7%);
- анальгетикам и нестероидным противовоспалительным препаратам (5,8%).
Лидерами среди терапевтических областей локальных исследований и исследований биоэквивалентности дженериков и биоаналогов отечественных спонсоров являются :
- неврология (12,8%);
- онкология (9,4%);
- кардиология и сердечно-сосудистые заболевания (9,4%);
- эндокринология (8,1%);
- ВИЧ и туберкулез (8,1%).
Обзор
Морфин, героин, эфедрин, мышьяк и свинец — не так давно эти вещества продавались как лекарства и назначались в качестве средств от кашля, боли или воспаления. Тестировать их сразу in vivo пришлось детям, беременным женщинам и остальным пациентам. О побочных эффектах тогда задумывались мало, альтернативных лекарств не было.
Сейчас от появления нового вещества, перспективного в плане решения каких-либо медицинских проблем до его регистрации в качестве лекарственного препарата проходит около 10-12 лет. За это время вещество подвергается сотням тестов и экспериментов. Но и после обретения торгового названия и своего места на аптечной полке лекарство многократно проверяют.