По характеру морфологических изменений патология называется гепатоцеребральной дистрофией, по авторам — болезнь Вильсона-Коновалова. Еще одно наименование — гепатолентикулярная дегенерация — уточняет и дополняет природу заболевания.
Современные исследования позволили причислить болезнь к врожденному нарушению метаболизма соединений меди с передачей по наследству. Выявлен ответственный за нее участок мутирующего гена. Несомненна связь с циррозом печени. Для болезни Вильсона-Коновалова характерно раннее выявление: признаки появляются у детей школьного возраста и младше.
История изучения
С конца XIX века врачи обращали внимание и описывали неясное заболевание, которой сопровождалось непроизвольными движениями в конечностях, в мышцах туловища, скованностью, нарушением речи, трудностями глотания пищи, реже — психическими расстройствами со снижением интеллекта, развитием слабоумия. Оно напоминало рассеянный склероз, поэтому называлось «псевдосклерозом».
Английский врач невропатолог С. Вильсон в 1912 году впервые связал симптоматику болезни с обязательным наличием цирротических изменений в печени, выявил взаимосвязь с поражением лентикулярных ядер головного мозга.
При этом отметил отсутствие изменений в пирамидном тракте больных (так именуются группа ядер и нервных путей человека, ответственных за движения). Значительный вклад в исследования по этой теме внес российский ученый академик Н.В. Коновалов.
Что известно о распространенности?
Статистика показывает, что болезнь Вильсона-Коновалова выявляется у трех человек на 100 тыс. населения. Показатель выше в тех странах, где возможны близкородственные браки. Согласно одним авторам — чаще болеют мужчины, другие утверждают, что одинаково восприимчивы как мужчины, так и женщины.
В семьях, имеющих носителей патологического генома, заболевают 25% братьев и сестер. При этом родители могут быть клинически здоровы. Симптомы начинают проявляться в среднем с 11 до 25 лет.
Каким образом наследуется болезнь?
Тип наследования аномального гена болезни называется аутосомно-рецессивным. Это означает, что заболеть могут только дети, получившие сразу два мутантных гена (от матери и отца). Родители являются гомозиготными носителями.
Если ребенок получает ген только от одного из родителей, то он не заболевает, но становится гетерозиготным носителем. У него возможны отклонения в обмене меди, но слабо выраженные. Насколько здорово будет третье поколение, зависит от встречи с аналогично измененной структурой хромосом жены или мужа.
Известен ген болезни — АТР7В — он обеспечивает синтез транспортирующего медь белка (церуллоплазмина). Находится на длинном плече в хромосоме №13, на участке с кодом 13q14-q21.

Мутации связаны с заменой последовательности включения аминокислот
Обнаружены разные виды мутаций у пациентов в Китае и странах Запада. Причины нарушений остаются неясными. У 10% больных вообще не выявлены генные изменения. Установлено, что в проявлении заболевания важное значение принадлежит факторам, поражающим печень. К ним относятся перенесенные инфекционные болезни (вирусный гепатит), интоксикация.
Роль меди в норме и при гепатолентикулярной дегенерации
Здоровый человек получает ежедневно с пищей 2 мг меди, из них усваивается только 1/3 часть, но и этого вполне хватает для обеспечения потребности организма.
Медь необходима печени:
- для синтеза белков и ферментов, а значит для роста, физического и умственного развития;
- образования гемоглобина, транспорта железа;
- синтеза эритроцитов и лейкоцитов;
- обеспечения эластичности стенок сосудов за счет участия в синтезе коллагена;
- стимуляции гормональной активности гипофиза и инсулина;
- выработки клеток иммунитета;
- образования антиоксидантов, предотвращения раннего старения.
 Признаки болезни печени
Признаки болезни печени
Попадая их тонкого кишечника в кровь, по воротной вене молекулы меди доставляются в печеночные клетки. Здесь часть связывается с белком церулоплазмином и возвращается в кровяное русло, другая (лишняя) — переходит в желчь и удаляется из организма. Одна молекула церуллоплазмина включает 8 атомов меди.
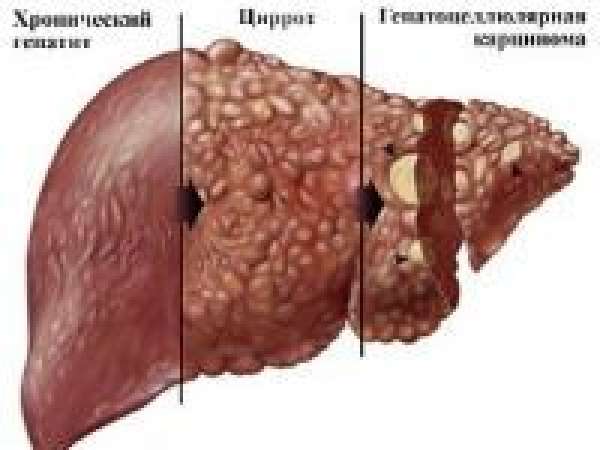
В патогенезе болезни Вильсона-Коновалова нарушаются обе функции. Церулоплазмин разрушается. Кровь больного переполняется значительным количеством несвязанной меди. Она откладывается в разных органах: в печени, головном мозге, почках, роговице глаз, переходит в мочу.
В гепатоцитах печени, как ответная реакция на внедрение меди образуется воспаление, переходящее в узловой цирроз. В почечной ткани страдают проксимальные канальцы. Повреждение головного мозга выражается в поражении базальных ганглиев, ядер мозжечка и черной субстанции.
В тканях глаза медь откладывается вокруг роговой оболочки и образует видимое кольцо Кайзера-Флейшера. Преимущественное отложение меди в одном из органов зависит от возраста. Для детей типично поражение печени. После 20 лет основные разрушения локализуются в головном мозге.
Какие изменения мозга вызывают признаки болезни?
Гепатоцеребральная дистрофия вызывает в тканях головного мозга размягчение нескольких ядерных образований (лентикулярного, хвостатого, зубчатого, подбугорных), мозжечка, глубинных слоев коркового слоя. В них образуются мелкие кисты.
Характерные токсические изменения различают:
- сосудистые — атония мелких сосудов способствует застойным явлениям крови, отеку окружающей ткани, гибели клеток, кровоизлияниям с выделением гемосидерина;
- клеточные — дистрофии подвергается макроглия нейронов, возможно формирование глии Альцгеймера, со временем клетки гибнут.
Установлено, что клеточные нарушения более распространены при позднем начале болезни и медленном течении. В печени атрофический процесс приводит к гибели гепатоцитов, целых долек с образованием некротизированных узлов. Между ними остаются очаги восстановления, нормальные клетки.
Образуются новые сосуды, которые формируют связи между ветками воротной вены печени и руслом нижней полой
Как проявляется заболевание?
Для гепатоцеребральной дистрофии характерно начало в детском возрасте или до 25 лет, дальнейшее развитие принимает хроническое течение с прогрессированием клинических проявлений. Часто до появления неврологических симптомов болезни Вильсона-Коновалова обнаруживают расстройства функций печени, кишечника.
У пациентов возникают тупые боли в подреберье справа, желтуха, понос или запор. Реже развивается гепатолиенальный синдром. Среди неврологических признаков:
- гиперкинезы (непроизвольные движения в руках и ногах, тремор), похожи на кривляние;
- мышечные спазмы (ригидность) и судороги;
- параличи;
- эпилептиформные припадки;
- нарушенная речь;
- слюнотечение;
- расстройства поведения, психики.
В отличие от ишемических нарушений и кровоизлияний нет нарушений чувствительности.
Среди прочих симптомов болезни, указывающих на поражение внутренних органов:
- кольцо Кайзера-Флейшера — зеленоватые отложения меди по окружности роговой оболочки, появляются на поздней стадии болезни;
- пигментные пятна на коже лица и туловища желтовато-коричневого цвета;
- признаки геморрагических нарушений — частые носовые кровотечения, кровоточивость десен, мраморный оттенок кожи, цианотичность губ, пальцев;
- боли в суставах;
- повышенная ломкость костей, переломы из-за остеопороза;
- профузное выделение пота.
Симптомы патологических нарушений в печени выявляются в 1/3 случаев, чаще их можно обнаружить в анализах крови по повышению уровня трансаминаз, билирубина, падению тромбоцитов и лейкоцитов, гемолитической анемии. Печень значительно увеличена.

Клинические обследования подтверждают активный гепатит с переходом в цирроз
Течение и формы болезни
По характеру течения болезни различают острую и хроническую формы. Острая — типична для раннего детского возраста, отличается молниеносным развитием, быстрым летальным исходом даже при лечении. Хроническая форма — протекает медленно с постепенным утяжелением неврологической симптоматики.
В зависимости от преобладающего поражения органов принято выделять пять разновидностей заболевания. Дрожательно-ригидная — встречается наиболее часто, начинается в юношеском возрасте, протекает медленно с ремиссиями и обострениями.
Состояние ухудшается внезапно, незначительно повышается температура тела, в руках определяется сочетание ригидности мышц и ритмичных дрожательных подергиваний по 2–8 движений в секунду. Проявления усиливаются при мышечном напряжении, волнениях, а в спокойном состоянии, во сне исчезают.
Возможно нарушение глотания и речи, насильственные движения (атетоидные, аналогичные хорее). Пациенты живут в среднем 6 лет.
Дрожательная — возникает к 20 годам, отличается медленным течением. В клинике болезни преобладает дрожание рук. Мышечная ригидность появляется много лет спустя. Характерна потеря мимики лица, медленная речь без эмоций, иногда снижение тонуса мышц. Значительно страдает психика человека, возможны вспышки агрессии, эпилептиформные припадки. Длительность жизни пациентов до 15 лет и более.
Брюшная — наиболее тяжелый вариант течения болезни Вильсона-Коновалова у детей. Поражение печени с выраженной недостаточностью приводит к раннему смертельному исходу. Неврологическая симптоматика не успевает развиться. Продолжительность заболевания не превышает 5 лет, ребенок может погибнуть за несколько месяцев.
Ригидно-аритмогиперкинетическая — другое название — ранняя форма. Начинается в детстве, характерно быстро прогрессирующее течение. В клинике проявляется значительной мышечной ригидностью со стойкими контрактурами, замедленностью и трудностью передвижения, нарушенной речью и глотанием, частыми хореоатетоидными движениями. Интеллект снижается умеренно. Болезнь длится в течение двух-трех лет до гибели ребенка.
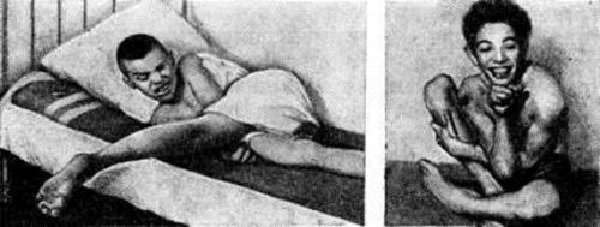
У пациентов возникает судорожный плач или смех
Экстрапирамидно-корковая — редкая форма течения болезни. К характерным признакам добавляются парезы и параличи, тяжелые припадки эпилепсии, развитие полного слабоумия. В коре полушарий мозга образуются обширные очаги размягчения. Средняя длительность — 6–8 лет. Неизбежен летальный исход.
Самая ранняя смертность у детей с брюшной формой. В половине случаев болезни наблюдается массивный некроз печени, гемолитические нарушения, желудочно-кишечное кровотечение (последствия портальной гипертензии). От неврологических изменений пациенты без лечения погибают спустя 5–15 лет.
Диагностика
Диагноз ставится на основании клинической симптоматики, сведений о заболеваемости в семьях родителей, заключения генетических исследований. В специализированных неврологических отделениях проверяют:
- количество меди в сыворотке крови;
- концентрацию церуллоплазмина (оба показателя при болезни снижаются);
- выделение меди с мочой (растет);
- осмотр окулиста с помощью щелевой лампы позволяет выявить кольца Кайзера-Флейшера, иногда вместо полного кольца обнаруживают «обломки».
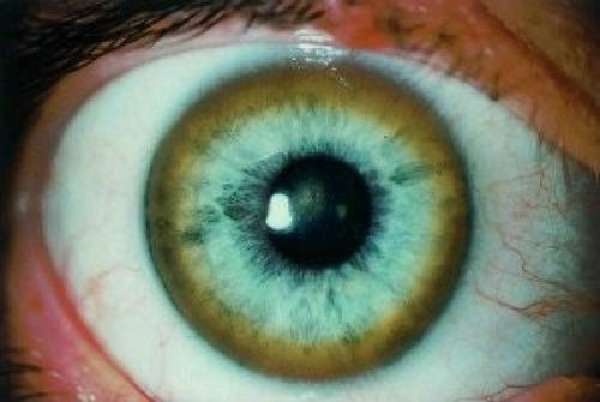
На фото из щелевой лампы хорошо видно зеленоватое кольцо, оно имеется не у всех пациентов
Имеют значение биохимические пробы печени, рост трансаминаз, щелочной фосфатазы, билирубина, тимоловой пробы, изменение белковых фракций, выявление сниженных показателей свертываемости.
Для общего анализа крови типично снижение эритроцитов и тромбоцитов, лейкопения. Почечные канальцевые изменения проявляются обнаружением в моче глюкозы, солей фосфорной кислоты, уратов, белка.
Компьютерная и магниторезонансная томография черепа выявляет изменения мозга до появления характерных неврологических симптомов: увеличение (расширение) желудочков, очаговые нарушения в зоне таламуса, скорлупы и бледного шара. В сложных случаях проводят биопсию печени, определяют содержание меди в гепатоцитах.
Для генетических исследований болезни Вильсона-Коновалова используют ДНК-маркеры, которые обладают высокой точностью. В дифференциальной диагностике учитывают, что церулоплазмин снижается при гепатитах, перенесенном голодании.
Возможности лечения
Приведенные сроки продолжительности жизни пациентов при современном уровне медицины удается значительно увеличить при помощи раннего начала лечения болезни Вильсона-Коновалова и использования всех возможных средств.
Питание пациента должно проводиться согласно диетическому столу №5а (для заболеваний печени). Исключаются все острые блюда, жареные и жирные. Одновременно для ограничения поступления меди с пищей категорически запрещаются: изделия и натуральные продукты, содержащие шоколад, орехи, кофе, мясо печени, раков и кальмаров, сухофрукты, бобовые, крупы из цельной пшеницы.
Основное направление — обеспечение вывода излишков меди. Главным препаратом считается D-пеницилламин. Препарат назначается с небольшой дозы по схеме, затем она увеличивается. Необходимо учитывать пожизненный прием лекарства и его негативные свойства.
Специалисты обращают внимание на низкое качество отечественного Пеницилламина, его токсичность. У некоторых пациентов с гепатолентикулярной дегенерацией побочные явления заключаются в дерматитах, анемии.
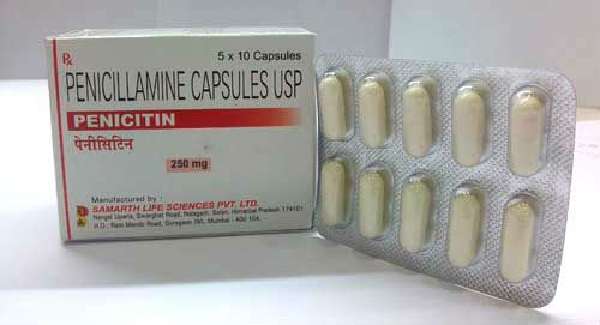
Препарат образует с медью комплексы, которые переходят в мочу и выделяются из организма, кроме того, способен подавлять активность внутриклеточной меди
Другое направление — терапия солями цинка (сульфат, оксид). Применение комбинаций D-пеницилламина с препаратами цинка позволяет проводить терапию низкими дозами и избегать отрицательных последствий. Если заболевание выявлено до начала клинических проявлений, то рекомендуются только препараты цинка. Дополнением является использование Унитиола.
В обязательном порядке пациенту назначаются гепатопротекторные средства: витамины, Эссенциале. Для поддержки проводимости в нервных волокнах необходимы витамины группы В. Предложен способ био-гемоперфузии крови с живыми изолированными клетками печени и селезенки. Методику называют «вспомогательной печенью».
Она проводится в специализированных центрах при безуспешной терапии для поддержки организма пациента до операции трансплантации печени. Пересадка здоровой печени донора помогает решить множество проблем лечения.
Сама операция требует подготовки больного, учета рисков в связи со сниженной свертываемостью крови. Это наиболее радикальный метод лечения форм болезни с преобладающим поражением печени.
Магнито-резонансное обследование применяют в процессе контроля за лечением
Эффективность лечения значительно улучшается при ранней диагностике. Удается достичь уменьшения проявлений. Пациенты остаются социально адаптированными людьми: способны полностью к самообслуживанию, учатся, работают по профессии, создают семью. Имеются наблюдения за молодыми женщинами с болезнью Вильсона-Коновалова, выносившими беременность и родившими здоровых детей.
Раз эффект лечения болезни зависит от раннего выявления и начала, то семьям, в которых есть ребенок с гепатолентикулярной дегенерацией, необходимо обследовать его братьев и сестер с применением современных молекулярно-генетических методик. Знание будущих родителей о своем носительстве предотвратит рождение больного малыша.